
fasta格式
fasta:一种基于文本用于表示核酸序列或多肽序列的格式。缩写为 fa
特征: 两部分, id行和序列行
- – id行:以“>”开头, 有时候会包含注释信息,如 chr1、chr2 …
- – 序列行:一个字母表示一个碱基/氨基酸,ATCGN 或 20种氨基酸
fastq格式
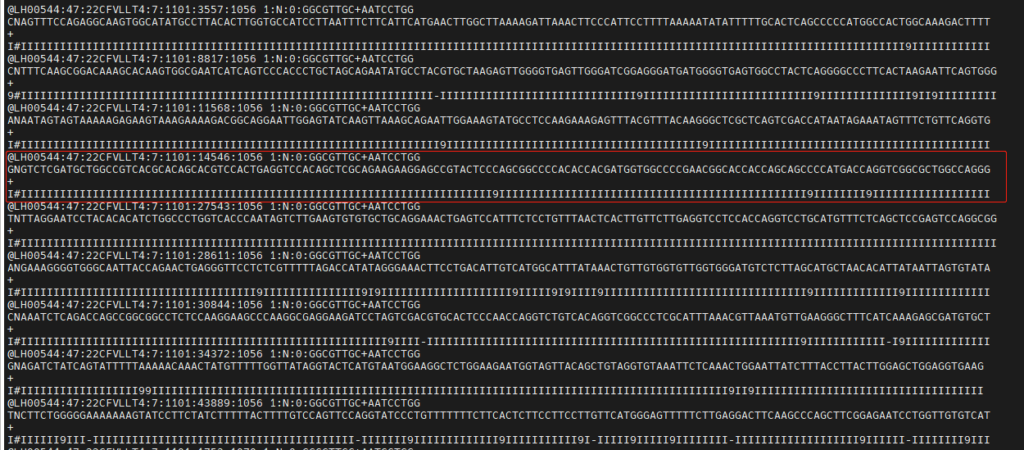
fastq:一种保存生物序列(通常为核酸序列)及其测序质量得分信息的 文本格式。缩写为 fq
FASTQ文件中,一个序列通常由四行组成,一般是生信分析的起点,一般拿到手的数据即fastq数据(少数包lan测序可能会拿到bcl数据,需要用index配合bcl2fastq软件转换成fastq数据。)
- 第一行:以 @ 开头,之后为序列的标识符以及描述信息
- 第二行:为序列信息,如 ATCG
- 第三行:以 + 开头,之后可以再次加上序列的标识及描述信息(保留行)
- 第四行:为碱基质量值,与第二行的序列相对应,长度必须与第二行相同
gff格式>f格式
GFF和GTF是两种最常用的基因组注释格式,在信息分析中建库时除了需要fasta文件一般还会需要这两种文件,提取需要的信息进行注释。
GFF
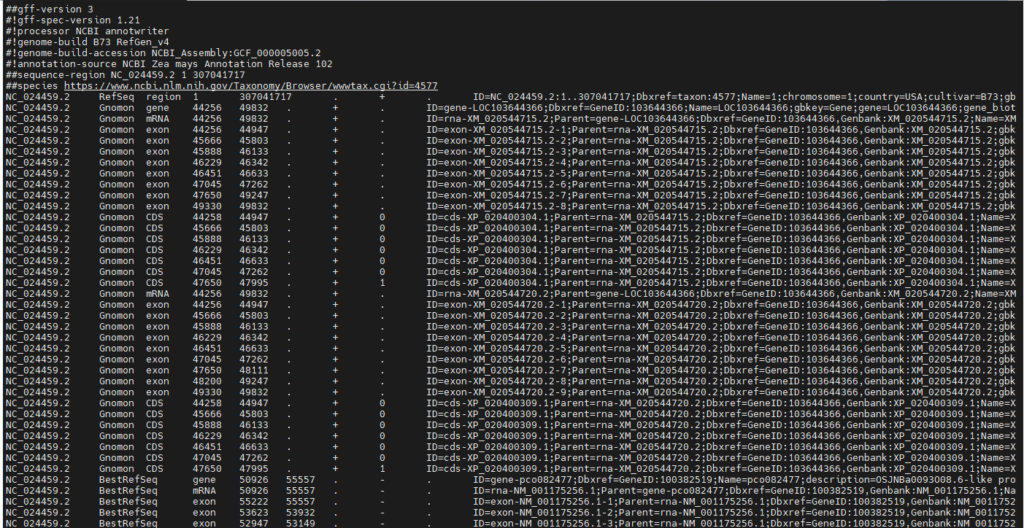
GFF(General Feature Format)是一种用来描述基因组特征的文件,现在我们所使用的大部分都是第三版(gff3)。
gff文件除gff1以外均由9列数据组成,前8列在gff的3个版本中信息都是相同的,只是名称不同(现在一般只使用gff3版本,这里只介绍gff3):
seqid:参考序列的id。source:注释的来源。如果未知,则用点(.)代替。一般指明产生此gff3文件的软件或方法。type: 类型,此处的名词是相对自由的,建议使用符合SO惯例的名称(sequenceontology),如gene,repeat_region,exon,CDS,UTR等。start:开始位点,从1开始计数(区别于bed文件从0开始计数)。end:结束位点。score:得分,对于一些可以量化的属性,可以在此设置一个数值以表示程度的不同。如果为空,用点(.)代替。strand:“+”表示正链,“-”表示负链,“.”表示不需要指定正负链。phase:步进。对于编码蛋白质的CDS来说,本列指定下一个密码子开始的位置。可以是0、1或2,表示到达下一个密码子需要跳过的碱基个数。attributes:属性。一个包含众多属性的列表,格式为“标签=值”(tag=value),不同属性之间以分号相隔。
GTF
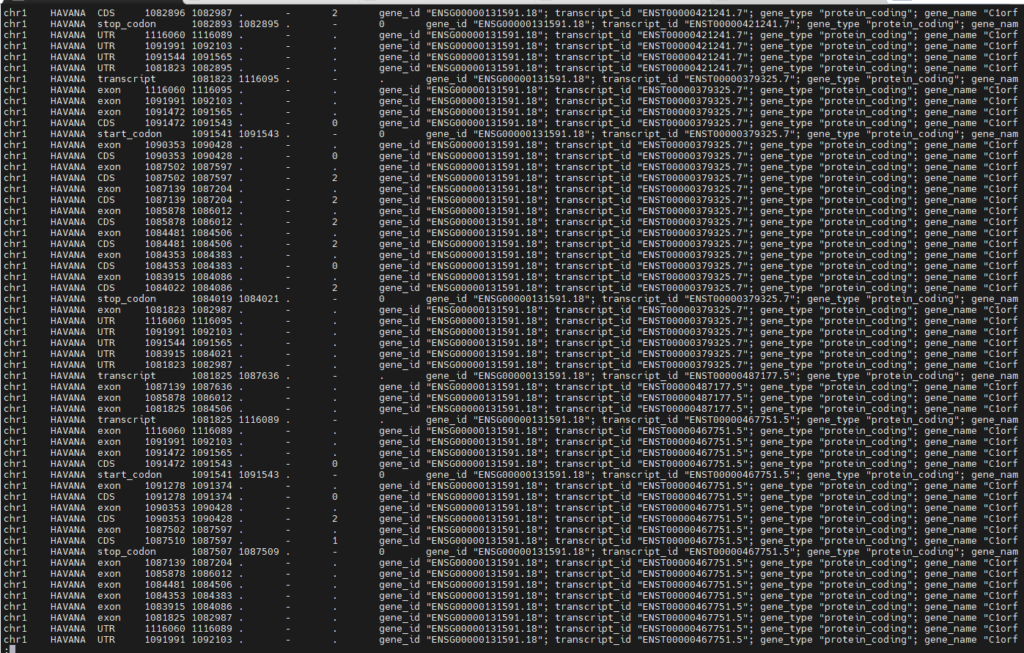
gtf全称为gene transfer format,主要是用来对基因进行注释,当前所广泛使用的gtf格式为第二版(gtf2)。以下均基于gtf2叙述:
seqname: 序列的名字。通常格式染色体ID或是contig ID。-
source:注释的来源。通常是预测软件名或是公共数据库。 feature:基因结构。CDS,start_codon,stop_codon是一定要含有的类型。start:开始位点,从1开始计数。end:结束位点。score:这一列的值表示对该类型存在性和其坐标的可信度,不是必须的,可以用点“.”代替。strand:链的正向与负向,分别用加号+和减号-表示。frame:密码子偏移,可以是0、1或2。attributes:必须要有以下两个值:
gene_id value; 表示转录本在基因组上的基因座的唯一的ID。gene_id与value值用空格分开,如果值为空,则表示没有对应的基因。
transcript_id value; 预测的转录本的唯一ID。transcript_id与value值用空格分开,空表示没有转录本。
GTF与GFF比较
GFF全称为general feature format,这种格式主要是用来注释基因组。GTF全称为gene transfer format, 这种格式主要是用来对基因进行注释。
GTF 的第九列,通常为:
gene _ id "At1ge0001"; transcript _ id "At1g0ee01.1";......而 GFF 的第九列,通常为:
ID =mrnae01; Name = abc
ID =exon1; Parent =mrnae01
ID =exon2; Parent =mrnae01GTF与GFF互相转换
gffread、GenomeTools、AGAT (Another GTF/GFF Analysis Toolkit)等工具都可以实现GFF和GTF的转换。
这里只介绍gffread工具:
gffread不仅可以实现GTF与GFF的互相转换,而且还可以对GFF文件进行过滤处理。可以直接读取GTF文件。
用法:
Usage:
gffread <input_gff> [-g <genomic_seqs_fasta> | <dir>][-s <seq_info.fsize>]
[-o <outfile.gff>] [-t <tname>] [-r [[<strand>]<chr>:]<start>..<end> [-R]]
[-CTVNJMKQAFGUBHZWTOLE] [-w <exons.fa>] [-x <cds.fa>] [-y <tr_cds.fa>]
[-i <maxintron>]
Filters and/or converts GFF3/GTF2 records.
<input_gff> is a GFF file, use '-' if the GFF records will be given at stdin-g 序列文件,即GFF/GTF文件第一列ID对应的序列文件。
-i 丢弃掉内含子大于的转录本(mRNA/transcript)
-r 起始和终止位置,填写示例100.10000即为输出与100到10000有重叠的所有转录组,也可以限制序列ID及链,填写示例:+Chr1:100..10000。
-R 丢弃掉此范围的转录本,与-r相反。
-U 丢弃掉 single-exon的转录本
-C 丢低调无CDS的转录本。
-V 丢弃掉含有移码突变的转录本。
-H 如果使用了-V,则重新检查并调整内含子相位,避免由于翻译起始位点选择的位置不对导致移码突变的产生。
-B 如果使用了-V, 对于单外显子基因,则重新检查相反的链,是否存在移码突变。
-N 丢弃掉多外显子基因剪接位点不是常见的 GT-AG, GC-AG or AT-AC序列。
-J 丢弃掉没有起始密码子或者终止密码子的转录本,仅保留有完整编码框的转录本。
--no-pseudo: 过滤掉含有 'pseudo' 的注释信息
-M/--merge : 合并完全相同的或者存在包含关系的转录本。
-d:使用 -M 将合并信息输出到文件中
--cluster-only: 类似于 --merge 但是不合并转录本
-K 对于-M 选项:also collapse shorter, fully contained transcripts
with fewer introns than the container
-Q 对于-M 选项:移除包含关系的转录本的限制条件:多外显子转录本将会合并,如果他们内含子位置完全一样,单外显子转录本只需要有80%一样即可合并。
--force-exons: 使GFF features的最小层级为exon
-E 对于重复的 ID或者 GFF/GTF 其他潜在的格式问题给出警告信息。
-Z 将内含子小于4 bp的邻近的两个外显子合并为一个。
-w 输出每个转录本的外显子序列
-x 输出CDS序列
-W 对于 -w 和 -x 选项,输出外显子位置坐标到FASTA序列的ID中
-y 输出蛋白质序列
-L 将Ensembl GTF 转换为 GFF3 conversion (implies -F; should be used with -m)
-o 输出"filtered" 后的GFF文件 。
-T -o 参数将输出 GTF格式。GFF转换GTF
gffread input.gff3 -T -o out.gtfGTF转换GFF3
gffread input.gtf -o out.gff3根据GFF或者GTF提取CDS,蛋白质和外显子序列
gffread gene.gff3 -g genome.fa -x cds.fa -y pep.fa -w cdna.fa只提取翻译后蛋白序列
gffread gencode.v19.annotation.gff3 -g hg19.fa -y tr_pep.fa根据reference提取CDS序列
gffread gencode.v19.annotation.gff3 -g hg19.fa -x cds.fabed格式
bed文件一般代表区域信息,如表示peak位置的bed文件,表示基因注释的bed12文件。
bed文件每一行对应信息
- 必须包含的3列信息:
- chrom:染色体名字 (e.g:chr3,chrY,chr2_random或者scaffold671)。
- chromStart:基因在染色体或scaffold上的起始位置(0-based)。
- chromEnd:基因在染色体或scaffold上的终止位置 (前闭后开)。
- 可选的9列信息:
- name:bed文件的行名。
- score:本条基因在注释数据集文件中的评分(0-1000),在Genome Browser中会根据不同区段的评分显示对应的阴影强度(评分越高灰度越高)。
- strand:链的方向+、-或. (.表示不确定链的方向)
- thickStart:CDS区(编码区)的起始位置,即起始密码子的位置。
- thickEnd: CDS区 (编码区)的终止位置,即终止密码子的位置。
- itemRgb:RGB颜色值(如:255,0,0),方便在GenomeBrowser中查看。
- blockCount:bed行中外显子的数目。
- blockSizes:逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的碱基数。
- blockStarts:逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的起始位置(数值是相对ChromStart计算的)。
注:bed文件只有前三列格式是一定的,后面的内容信息可以自由确定。
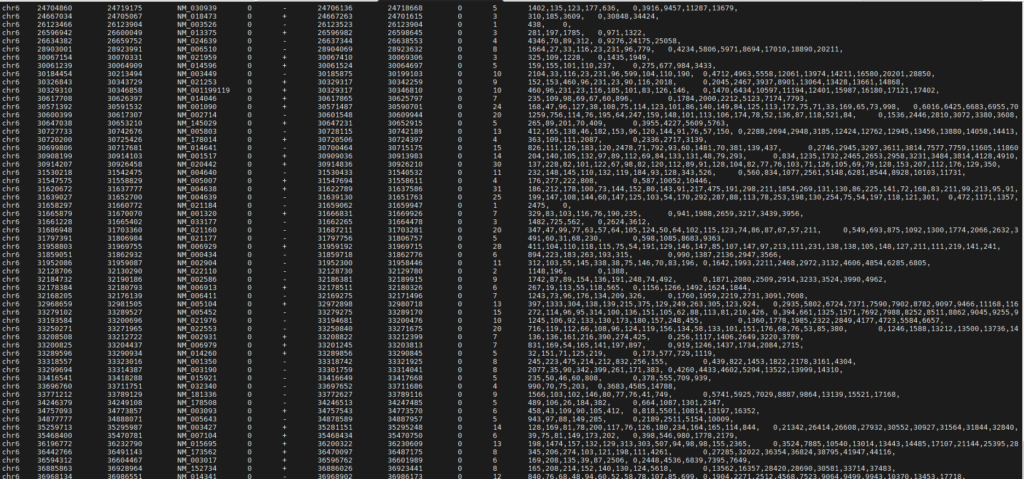
表示基因注释时,gtf/gff和bed文件的区别:
- gtf/gff文件一行表示一个exon/CDS等子区域,多行联合表示一个gene;bed文件一行表示一个gene;
- gtf文件中碱基位置定位方式是1-based(即起始的碱基记为1),而bed中碱基定位方式是0-based(即起始的碱基记为0)
sam/bam格式
sam文件全称The Sequencing Alignment/Map Format,是Alignment/Map步骤(比对)bwa/STAR/HISAT2等软件对结果的标准输出文件,用于存储reads比对到参考基因组的比对结果,是一个纯文本格式,文件一般较大。为了节省硬盘存储,一般使用其高效压缩的二进制格式bam文件。
sam:reads比对到参考基因组上的比对结果文件
bam:sam文件的二进制版本
一般对于sam/bam文件的查看处理都需要通过samtools软件来实现。(以后会专门出一期文章来介绍samtools软件)
bam文件一般包括两个部分
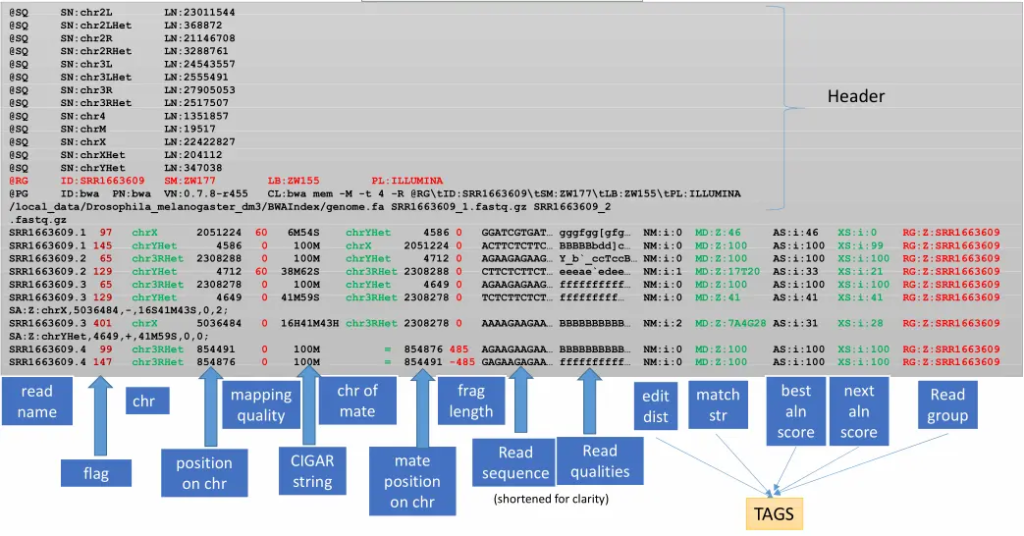
header内容
- @HD:是必须的标准文件头,包含版本信息;
- @SQ:参考序列染色体名字和长度信息 (SN:scaffold name; LN: length);
- @RG:重要read group信息,通常包含测序平台,测序文库和样本ID等信息,分析时用于区分不同样本(重测序时用到);
- @PG:生成此文件的操作过程和参数信息 (program)。
record内容
- read名称;
- 比对信息位flag值;
- 参考序列染色体编号;
- 5′端起始位置;
- MAPQ:mapping quality,描述比对的质量,数字越大,特异性越高;
- CIGAR字符串,记录插入、删除、错配等信息;
- 配对read所比对到的染色体,仅双端测序的数据才有;
- 配对read所比对到的位置,仅双端测序的数据才有;
- 插入片段的长度,仅双端测序的数据才有;
- read序列;
- read质量值;
- 12列以后的信息都是metadata,程序用标记
其中第二列的flag值是一个非常重要的参数:
flag一共有12个标签,使用16进制数表示,每个标签值是2^(n-1),其中n<=12,每个值有其对应的唯一解释含义,SAM Format有对每个flag值的具体解释
bam文件的操作前提需要创建索引:
samtools index ***.bam #可选择索引类型Wiggle、bigwig和bedgraph文件
wig、bigwig和bedgraph是UCSC规定的一些文档格式,可以无缝连接到UCSC的Genome Browser工具里面进行可视化。
上述bam和sam文件可以帮助我们探索reads在参考基因组中的比对情况,比对结果是文本文件,不方便阅读。而wiggle(简称wig)、bigwig(简写bw)以及bedgraph(简写bdg)只包含区域和区域的覆盖度信息,文件更小,用于可视化更方便,可以导入基因组浏览器(Genome Browser)中进行可视化,以查看reads在参考基因组各个区域的覆盖度并检测测序深度。
下面是UCSC官网给出的wig、bigwig、bedgraph帮助文本
Wiggle Track Format :Genome Browser Wiggle Track Format (ucsc.edu)
bigWig Track Format :Genome Browser bigWig Format (ucsc.edu)
BedGraph Track Format :Genome Browser bedGraph Track Format (ucsc.edu)
tips: SOPs/coordinates – BaRC Wiki (mit.edu) 提供了这几种数据格式的构造及转换脚本
Wiggle/Bigwig
WIG文件的基本结构
WIG文件由一个或多个“记录”组成,每个记录描述一个连续测量值。记录由以下字段组成:
- 染色体名称(Reference Name):描述测量值所在的染色体的名称或编号。
- 起始位置(Start):测量值的起始坐标,通常是以0-based的方式表示。
- 结束位置(End):测量值的结束坐标,也是0-based。
- 测量值(Value):测量值的实际值。
BIGWIG文件是WIG文件的一种变体,它使用了更高效的压缩算法来存储数据。
BIGWIG文件的基本结构
BIGWIG文件由一个或多个“块”组成,每个块描述一个连续测量值。块由以下字段组成:
- 染色体名称(Reference Name):描述测量值所在的染色体的名称或编号。
- 起始位置(Start):测量值的起始坐标,通常是以0-based的方式表示。
- 结束位置(End):测量值的结束坐标,也是0-based。
- 测量值(Value):测量值的实际值。
- 块大小(BlockSize):块的大小,以字节为单位。
bedgraph
BEDGRAPH文件是BIGWIG文件的另一种变体,它使用了更简单的格式来存储数据。
BEDGRAPH文件的基本结构
BEDGRAPH文件由一个或多个“记录”组成,每个记录描述一个连续测量值。记录由以下字段组成:
- 染色体名称(Reference Name):描述测量值所在的染色体的名称或编号。
- 起始位置(Start):测量值的起始坐标,通常是以0-based的方式表示。
- 结束位置(End):测量值的结束坐标,也是0-based。
- 测量值(Value):测量值的实际值。
VCF文件格式
VCF(Variant Call Format)文件是一种用于存储基因组变异数据的文本格式,通常用于描述DNA或RNA测序数据中的单核苷酸变异和结构变异。VCF文件包括了各种类型的变异信息,如单核苷酸多态性(SNP)、插入/删除(Indel)、复合杂变异等。
文件后缀:vcf
文件元数据(File Metadata):VCF文件的开头通常包括一些元数据信息,用于描述文件的属性和来源,以及变异数据的版本等。元数据行以”##”开头,可以包括信息字段、格式字段、命令信息和样本信息字段等。
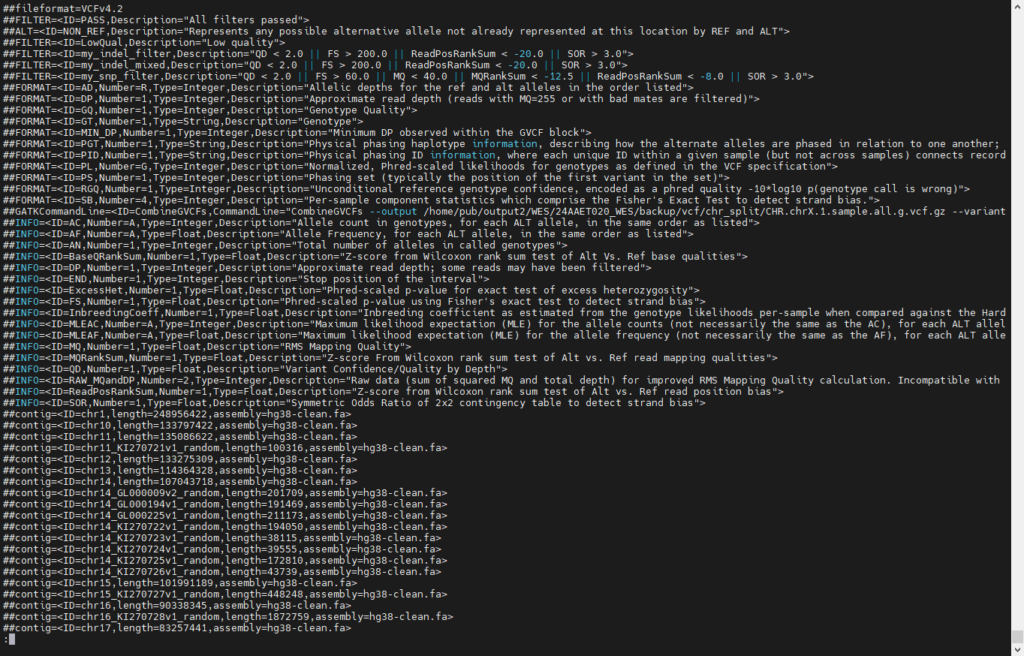
列名(Column Headers):VCF文件的列名行包括了各个字段的名称,通常以”#“开头,后跟字段名称,包括”CHROM”(染色体名称)、“POS”(变异位置)、“ID”(变异标识符)、“REF”(参考碱基)、“ALT”(变异碱基)、“QUAL”(质量分数)、“FILTER”(过滤信息)、“INFO”(变异信息)和”FORMAT”(样本格式)等。
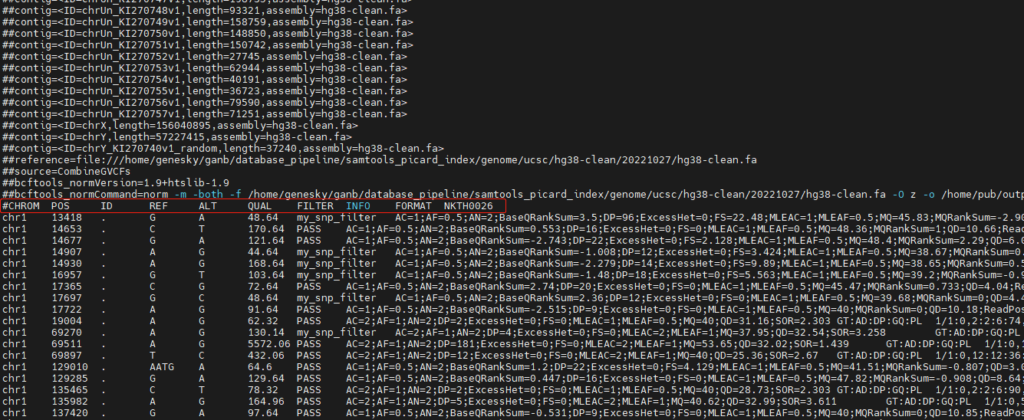
变异记录行(Variant Records):VCF文件的主体部分包括一行行的变异记录,每一行描述一个变异事件。变异记录行包括了各种字段,这些字段用制表符(“\t”)分隔,包括:
- CHROM:变异位点所在的染色体名称。
- POS:变异位点在染色体上的位置。
- ID:变异的唯一标识符。(一般为rs号,或者用’.’占位)
- REF:参考碱基。(参考基因组上该位置的碱基)
- ALT:变异碱基。(检测结果该位置的突变碱基)
- QUAL:质量分数,表示变异的质量估计。
- FILTER:过滤信息,描述变异是否通过了一些质量过滤。
- INFO:包含有关变异的详细信息的字段。
- FORMAT:描述每个样本中的数据格式。
- 样本数据:每个样本的具体数据,包括基因型信息、深度信息、质量信息等。(上图中样本名为“NKTH0026”)
VCF文件是用于存储和共享基因组变异信息的标准格式,在基因组学研究和生物信息学分析中广泛使用。它允许研究人员记录和交换关于基因组中的变异的重要信息,以便进一步的研究和解释。VCF文件的格式规范有不同的版本,因此要确保正确解析和处理VCF文件,最好查阅文件的元数据信息以了解其格式版本。
这里重点介绍一下VCF文件里的‘FORMAT’格式:
一般在文件的文件元数据中会有对本文本‘FORMAT’的介绍,只有结合FORMAT加上每个样本的数据才能够读懂VCF中存储数据的含义。
##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allelic depths for the ref and alt alleles in the order listed">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth (reads with MQ=255 or with bad mates are filtered)">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=MIN_DP,Number=1,Type=Integer,Description="Minimum DP observed within the GVCF block">
##FORMAT=<ID=PGT,Number=1,Type=String,Description="Physical phasing haplotype information, describing how the alternate alleles arephased in relation to one another; will always be heterozygous and is not intended to describe called alleles">
##FORMAT=<ID=PID,Number=1,Type=String,Description="Physical phasing ID information, where each unique ID within a given sample (but not across samples) connects records within a phasing group">
##FORMAT=<ID=PL,Number=G,Type=Integer,Description="Normalized, Phred-scaled likelihoods for genotypes as defined in the VCF specification">
##FORMAT=<ID=PS,Number=1,Type=Integer,Description="Phasing set (typically the position of the first variant in the set)">
##FORMAT=<ID=RGQ,Number=1,Type=Integer,Description="Unconditional reference genotype confidence, encoded as a phred quality -10*log10 p(genotype call is wrong)">
##FORMAT=<ID=SB,Number=4,Type=Integer,Description="Per-sample component statistics which comprise the Fisher's Exact Test to detect strand bias.">
#CHROM POS ID REF ALT......FORMAT NKTH0026
chr1 14677 . G A ......GT:AD:DP:GQ:PL 0/1:86,8:94:56:56,0,2693
chr1 129285 . G A ......GT:AD:DP:GQ:PL 0/1:14,6:20:99:129,0,544
chr1 188118 . C T ......GT:AD:DP:GQ:PGT:PID:PL:PS 1|1:0,11:11:33:1|1:2467680_A_G:414,33,0:2467680
chr1 827209 . G C ......GT:AD:DP:GQ:PL 1/1:1,250:251:99:9137,744,0
....................................................................................VCF文件一般是比对后的bam结合物种参考基因组文件(**.fa)、目标区域文件(**.bed),使用GATK工具包获得的结果。(以后会专门出一期文章来介绍GATK软件)
一般对于vcf文件的查看处理都需要通过bcftools软件来实现。(以后会专门出一期文章来介绍bcftools软件)
VCF文件操作前提是需要索引文件(VCF文件只能压缩后再建立索引)
bcftools index ***.vcf.gz #可选择索引类型
tabix ***.vcf.gz #可选择索引类型 (tabix对GFF、VCF、BED等文本都可建立索引,但必须是压缩后的文件)VCF&gVCF
gvcf文件与vcf文件都是vcf文件,不同之处在于gvcf文件会记录更多的信息,这里更多的信息指的是未突变的位点的覆盖情况,从下面的图我们可以直观的看出两者的区别
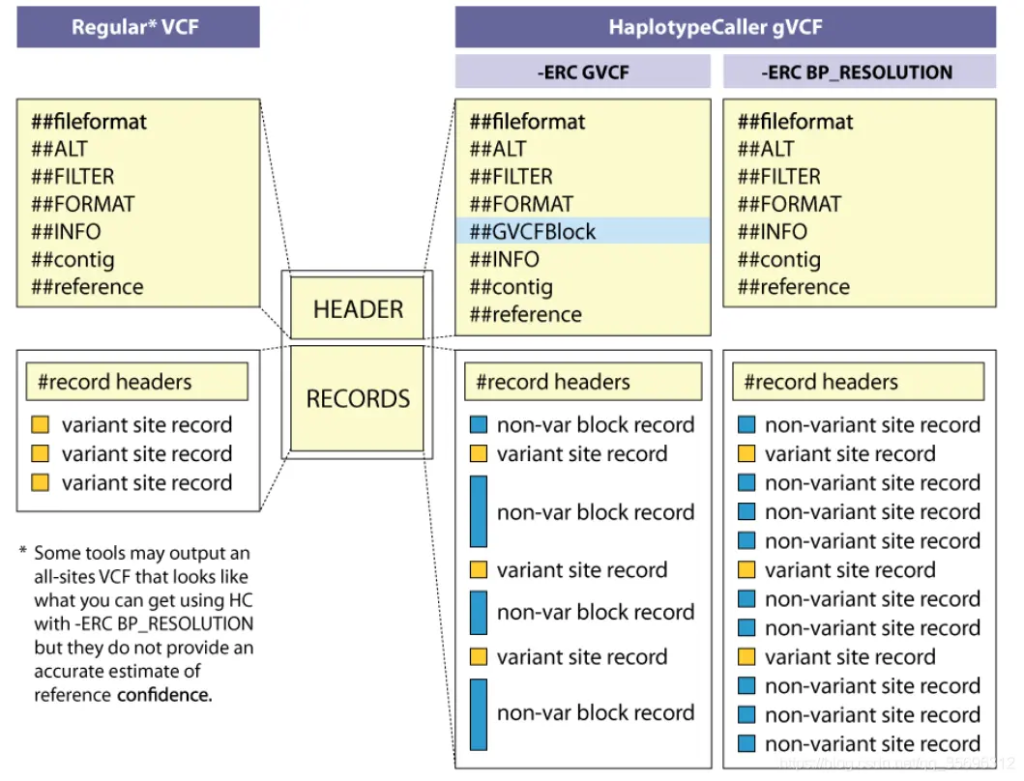
可以看到,gvcf文件也分两种,一种是-erc gvcf ,另一种是 -erc bp_resolution。这两种gvcf文件的区别在于前一种gvcf文件记录非突变位点的时候,以块的形式来记录;而后一种gvcf文件则是对非突变和突变位点一视同仁,前一种方式是为了有效的压缩文件的行数和大小,对后续的分析没有影响。一般推荐使用-erc gvcf 模式。
- VCF和VCF的最大区别是在于GVCF文件会记录所有的点,包括哪些没有突变的点。
在GVCF模式下,那些没有变异的点会形成一个未变异块,non-var block record。
gVCF的优点:能把多个样本的gVCF文件合并起来,方便后续的下游分析。
tips:VCF文件常见的都是压缩文件格式:sample.vcf.gz
这里要注意VCF文件的压缩要使用bgzip 命令;解压使用bgzip -d (普通的gzip命令不适用)
Plink文件格式
Plink文件格式是由Plink软件 所指定的存储变异数据的文本格式,和VCF文件存储的信息基本相同,可以实现互相转换。
Plink常用的文件格式有两套:map/ped 和 bim/fam/bed()。两组文件均没有列名,且每一列表示的意思是一定的。几种格式之间可以相互转换。在软件使用中推荐使用bim/fam/bed这种格式,读取速度快。
map/ped 文件
.map文件
map格式的文件, 主要是图谱文件信息, 主要包括染色体名称, 所在的染色体和所在染色体的坐标。
map文件包括:
- 第一列:染色体编号(1-22, X, Y or 0 if unplaced), 未知为0;
- 第二列:SNP名称(字符或数字或rs编号), 如果不重要, 可以从1编号, 注意要和bed文件SNP列一一对应;
- 第三列:染色体的摩尔位置(可选项, 可以用0);
- 第四列:SNP物理坐标;
.ped文件
ped格式的文件, 主要包括SNP的信息, 包括个体ID, 系谱信息, 表型和SNP的分型信息。
.ped 文件主要有 6 列,后面都是基因型:
- 第一列: Family ID # 如果没有, 可以用个体ID代替;
- 第二列: Individual ID # 个体ID编号;
- 第三列: Paternal ID # 父本编号;
- 第四列: Maternal ID # 母本编号;
- 第五列: Sex (1=male; 2=female; other=unknown) # 性别, 如果未知, 用0表示;
- 第六列: Phenotype (0=unknown; 1=unaffected; 2=affected) # 表型数据, 如果未知, 用0表示;
- 第七列以后: 为SNP分型数据, 可以是AT CG或11 12, 或者A T C G或1 1 2 2;
bim/fam/bed文件
.bim文件
bim文件存储每个遗传变异(通常是SNP)的相关信息,每一行代表一个遗传变异,共6列:
- 第一列:染色体编号(常用整数标记,如22表示第22条染色体,性染色体和线粒体染色体用’X’/‘Y’/‘XY’/‘MT’表示,而’0’ 代表染色体信息缺失);
- 第二列:变异标识符,这个就相当与每一个遗传变异的编号,常见的SNP可以采用以“rs”开头的编号;
- 第三列:每个遗传变异在基因组上的位置,用摩尔根或者厘摩尔根表示;
- 第四列:碱基对的坐标;
- 第五列:等位基因1(A1),通常是次要等位基因(minor allele);
- 第六列:等位基因2(A2),通常是主要等位基因(major allele)。
.fam文件
fam存储的是样本家系等信息,共6列:
- 第一列:家系编号(‘FID’);
- 第二列:个体编号(‘IID’; 不能是 ‘0’);
- 第三列:父系编号 (‘0’ 表示父系信息缺失);
- 第四列:母系编号(‘0’ 表示母系信息缺失);
- 第五列:性别编号(‘1’ = 男, ‘2’ = 女, ‘0’ = 性别未知);
- 第六列:表型值 (‘1’ = 对照, ‘2’ = 病例, ‘-9’/‘0’/表示表型缺失)。
.bed文件
bed存储基因型信息,是plink中的二元等位基因表。是一个二进制文本,存储的是每个样本在每个位点上的分型信息。(和表示区域信息的bed文件格式没有关系)
plink格式文件的相互转换
#1. bed/bim/fam 转为 ped/map
#input files: test.bed; test.bim; test.fam
#output files: test1.ped; test1.map
plink --noweb --file test --recode --out test1
#########################################################################################################
#2. ped/map 转为二进制格式 bed/bim/fam
#input files: test.ped; test.map
#output files: test2.bed; test2.bim; test2.fam
plink --noweb --file test --make-bed --out test2VCF文件和Plink文件
VCF文件和Plink文件都是存储突变分型的文本,可实现两种文本之间相互转换(本文介绍Plink软件、vcftools软件 两种)。
(Plink软件一般用于位点、表型关联分析,以后会详细介绍。)
#vcf 转 ped/map
#input files: test.vcf;
#output files: test.ped; test.map
plink --vcf test.vcf --recode --out test # 当出现错误,无法读取chrom时,加 --allow-extra-chr,可以强制程序接受编号
vcftools --vcf test.vcf --plink --out test
#########################################################################################################
#vcf 转 bed/bim/fam
#input files: test.vcf;
#output files: test.bed; test.bim ;test.fam
plink --vcf test.vcf --make-bed --out test # 当出现错误,无法读取chrom时,加 --allow-extra-chr,可以强制程序接受编号
#########################################################################################################
#bed/bim/fam 转 vcf
#input files: test.bed; test.bim; test.fam
#output files: test.vcf
plink --bfile test --export vcf --out test
plink --bfile test --recode vcf-iid --out test