
DNA-seq最常见的就是WES还有WGS以及各种target检测,就是最常见的测序方式。本片将以WES(全外显子测序分析)为例来介绍一整套分析流程。
下面是一整套WES分析流程的思维导图,这次只介绍前半部分的数据处理部分与SNV分析部分
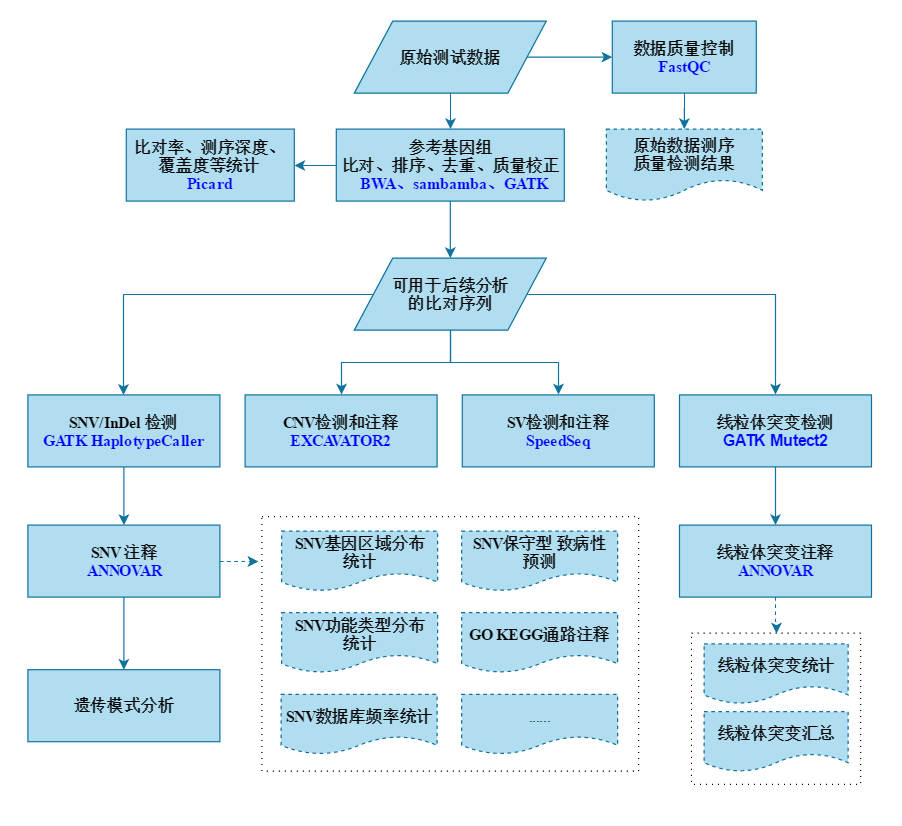
QC 数据质控
拿到数据的第一步就是QC,对原始数据经行质量统计
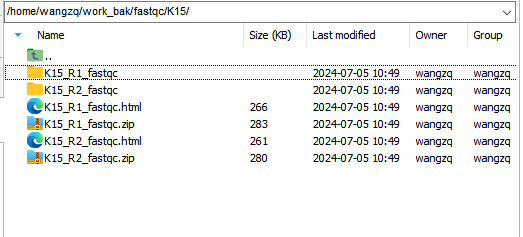
[wangzq@server6 fastqc]$ /home/software/fastqc/0.11.5/fastqc -t 10 --extract -q -o ./K15 --dir ~/tmp/ /home/project/K15_R1.fastq.gz /home/project/K15_R2.fastq.gz
Too many tiles (>500) so giving up trying to do per-tile qualities since we're probably parsing the file wrongly
Too many tiles (>500) so giving up trying to do per-tile qualities since we're probably parsing the file wrongly
# -t --threads 指定线程
# --extract 输出文件将在同一目录下进行解压缩,非交互式运行情况下建议添加此参数
# -q --quiet 禁止stdout上的所有进度消息,只报告错误。
# -o --outdir 指定输出文件路径
# -d --dir 用于生成报告图像时写入的临时文件。如果未指定,默认为系统临时目录。
运行结果:
包括了R1、R2端数据QC结果并整理出俩html报告
之后流程会进行一些可视化绘图,如:绘制错误率图、碱基占比图、碱基质量分布图…
Mapping 基因组比对(Alignment)
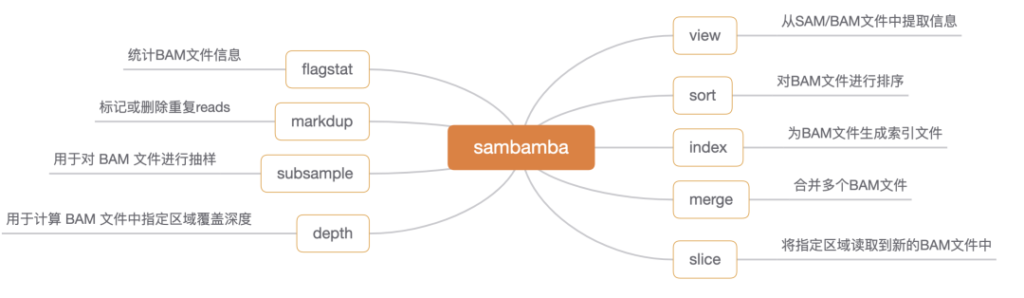
比对是指将我们的fastq数据比对到该物种的参考基因组上去,获得比对结果的bam/sam 文件。这里选择BWA软件mem算法来进行比对工作,使用samblaster、sambamba等工具过滤、标记来提高速度。
比对之前要准备该物种参考基因组的BWA索引(index)
sambamba重要用途:
/home/software/bwa/0.7.17/bwa mem -t 15 -k 32 -T 50 -B 8 -O 12 -L 10 -U 18 -R '@RG\tID:K15\tSM:K15\tLB:K15\tPL:illumina\\tPU:barcode' "/home/ucsc/hg38_rm_alt_genome/bwa_idx/hg38_modify.fa" /home/K15_R1.fastq.gz /home/K15_R2.fastq.gz |
/home/software/samblaster/0.1.24/samblaster --addMateTags --maxSplitCount 2 --minNonOverlap 20 --splitterFile ./K15/K15_sort.splitters.sam --discordantFile ./K15/K15_sort.discordants.sam 2> ./K15/K15.samblaster_run.log |
/home/software/sambamba/0.6.7/sambamba view -S -f bam -l 0 /dev/stdin |
/home/software/sambamba/0.6.7/sambamba sort -t 15 -m 6G --tmpdir=/home/tmp -o ./K15/K15_sort.bam /dev/stdin
#BWA mem:fastq序列比对参考基因组,生成比对结果文件bam/sam
# -t 指定线程
# -k 最小长度
# -T 输出的最低分数
# -B 不匹配的处罚
# -O 间隙开放对删除和插入的惩罚
# -L 5'-和3'-端部修剪的惩罚
# -U 未配对读取对的惩罚
# -R 读取组标题行 例如'@RG\tID:SampleName\tSM:SampleName\tLB:SampleName\tPL:illumina\\tPU:barcode'
# "/home/ucsc/hg38_rm_alt_genome/bwa_idx/hg38_modify.fa" 参考基因组索引文件
# K15_R1.fastq.gz K15_R2.fastq.gz 样本下机的fastq数据
# -o 结果sam文件输出路径(脚本中未指定,所以默认输出到stdout标准输出流,用管道符传输到下一个软件的输入去)
#
#samblaster:用于标记重复序列和选择输出reads不一致序列。
# -i --input 输入的sam文件(脚本中未指定,所以默认输入路径stdin,是前一个软件用管道符传输过来了的)
# --discordantFile 两端reads数据比对不一致的结果
# --splitterFile 比对结果低于指定参数的结果
# --addMateTags 将MC和MQ标记添加到所有输出配对的末端SAM行
# --maxSplitCount 最大拆分对齐数
# --minNonOverlap 最小重叠碱基数
# -o --output 结果sam文件输出路径(脚本中未指定,所以默认输出到stdout标准输出流,用管道符传输到下一个软件的输入去)
# 2> ./K15/K15.samblaster_run.log 把samblaster 软件运行结果简单汇总写在log中
#
#sambamba view:这里用的功能是sam格式转bam格式
# -S --sam-input 指定输入文件格式(默认为bam) 输入文件/dev/stdin 标准输入流
# -f --format 指定输出文件格式(默认是sam, 还支持bam, json, or msgpack )
# -l --compression-level 指定压缩级别(从0到9,仅适用于BAM输出)
# -o --output-filename 结果输出文件(脚本中未指定,所以默认输出到stdout标准输出流,用管道符传输到下一个软件的输入去)
#
#sambamba sort:这里是对bam结果排序
# -t 指定线程
# -m 指定最大内存memory
# --tmpdir= 用于存储中间文件的目录;默认为临时文件的系统目录
# -o 结果输出文件名
# <input.bam> 输入文件 一般写在命令最后面,这里用的是/dev/stdin标准输入流gawk 'BEGIN{OFS="\t"} {if($0~"@"){print;next} else {$10="*";$11="*";print} }' K15_sort.splitters.sam |/home/software/sambamba/0.6.7/sambamba view -S -f bam -l 0 /dev/stdin |/home/software/sambamba/0.6.7/sambamba sort -t 10 -m 1G --tmpdir=/home/tmp -o K15_sort.splitters.bam /dev/stdin
gawk 'BEGIN{OFS="\t"} {if($0~"@"){print;next} else {$10="*";$11="*";print} }' K15_sort.discordants.sam |/home/software/sambamba/0.6.7/sambamba view -S -f bam -l 0 /dev/stdin |/home/software/sambamba/0.6.7/sambamba sort -t 10 -m 1G --tmpdir=/home/tmp -o K15_sort.discordants.bam /dev/stdin
#分别对sort.splitters.sam sort.discordants.sam 文件进行简化处理(序列信息、碱基质量用*代替),并转换成bam格式。/home/software/sambamba/0.6.7/sambamba view view -f bam -F "not duplicate" -t 10 -o ./K15_sort_dup.bam K15_sort.bam
#对K15_sort.bam文件进行去重复(被flag标记的duplicate)samtools、sambamba、picard软件都可以实现去重复使用GATK对sort.bam结果进行下一步矫正处理
GATK (全称The Genome Analysis Toolkit)是用于二代重测序数据分析的一款软件,是基因分析的工具集。被广泛用于处理突变检测分析流程。gatk工具包里有很多内容,这里不做过多介绍。
GATK (broadinstitute.org)
$SOFT_GATK3 = "/home/software/java/1.8.0_181/bin/java -Djava.io.tmpdir=/home/tmp -jar -Xmx5g /home/software/gatk/3.5/GenomeAnalysisTK.jar";
$SOFT_GATK4 = "/home/software/java/1.8.0_181/bin/java -jar -Xmx8g /home/software/gatk/4.1.3.0/gatk-package-4.1.3.0-local.jar";
$SOFT_GATK3 -l INFO -T RealignerTargetCreator -R /home/ucsc/hg38_rm_alt_genome/hg38_modify.fa -I ./K15_sort_dup.bam -L /home/ucsc/hg38_rm_alt_genome/realign_Target_hg38.bed -o ./K15_sort_dup_realign.intervals --validation_strictness LENIENT
$SOFT_GATK3 -l INFO -T IndelRealigner -R /home/ucsc/hg38_rm_alt_genome/hg38_modify.fa -I ./K15_sort_dup.bam -L /home/ucsc/hg38_rm_alt_genome/realign_Target_hg38.bed -o ./K15_sort_dup_realign.bam --targetIntervals ./K15_sort_dup_realign.intervals --validation_strictness LENIENT
$SOFT_GATK4 BaseRecalibrator -R /home/ucsc/hg38_rm_alt_genome/hg38_modify.fa -I ./K15_sort_dup_realign.bam -O ./K15_sort_dup_realign_recalibrator.table $knownSitesDBsnp $knownSitesInDelGold $knownSitesInDel --tmp-dir /home/tmp
$SOFT_GATK4 ApplyBQSR -R /home/ucsc/hg38_rm_alt_genome/hg38_modify.fa -I ./K15_sort_dup_realign.bam -O ./K15_sort_dup_realign_recalibrator.bam --bqsr-recal-file ./K15_sort_dup_realign_recalibrator.table --tmp-dir /home/tmp
#这一块是使用GATK3,据数据库,对已知插入缺失位点校正,同时,只保留目标区域(本项目是外显子区域/home/ucsc/hg38_rm_alt_genome/realign_Target_hg38.bed)的数据,减少数据量
#这一部分只是对碱基质量的一些矫正,目标区域的保留,可以跳过,流程是能够跑通的。
mv ./K15_sort_dup_realign_recalibrator.bam ./K15_final.bam
mv ./K15_sort_dup_realign_recalibrator.bai ./K15_final.bai
K15_final.bam K15_final.bai 就是基因组比对所获得的最终结果,用于进行后续分析。
Mapping QC 质量统计
mapping qc即对mapping的结果进行整理汇总,对整个比对过程与结果给出一个可视化的统计分析。本步骤主要使用picard软件来实现。picard是基于java环境下的软件
Picard Tools – By Broad Institute
# 质量统计
$SOFT_PICARD = "/home/software/java/1.8.0_181/bin/java -jar -Xmx3g /home/software/picard/2.18.29/picard.jar";
$bam_rm_dup_flag =......#这里的bam_rm_dup_flag 是基于上面生成的K15_final.bam文件,去掉duplicate标记 的结果
$SOFT_PICARD CollectHsMetrics INPUT=$bam_rm_dup_flag OUTPUT=./K15.metrics R="/home/ucsc/hg38_rm_alt_genome/bwa_idx/hg38_modify.fa" BI=/home/ucsc/hg38_rm_alt_genome/Target_hg38.bed TI=/home/ucsc/hg38_rm_alt_genome/Target_hg38.bed PER_TARGET_COVERAGE=./K15.tag MINIMUM_MAPPING_QUALITY=0 MINIMUM_BASE_QUALITY=0 VALIDATION_STRINGENCY=LENIENT TMP_DIR=/home/tmp
#使用PICARD CollectHsMetrics (输入去掉dup标记的bam文件,参考物种fa,目标区域的bed),获得./K15.metrics,./K15.tag
$SOFT_PICARD CollectQualityYieldMetrics INPUT=$bam_rm_dup_flag OUTPUT=./K15.quality.metrics VALIDATION_STRINGENCY=LENIENT TMP_DIR=/home/tmp
#使用PICARD CollectQualityYieldMetrics(输入去掉dup标记的bam文件),获得./K15.quality.metrics
# 文库序列长度统计
$SOFT_PICARD CollectInsertSizeMetrics INPUT=$bam_rm_dup_flag O=./K15.insert H=./K15.insert.pdf VALIDATION_STRINGENCY=LENIENT TMP_DIR=/home/tmp
#使用PICARD CollectInsertSizeMetrics (输入去掉dup标记的bam文件),获得./K15.insert ./K15.insert.pdf生成gVCF文件
使用GATK4软件对比对结果单倍型召回,获得每个样本独立的gVCF文件。
# 执行GVCF
my $SOFT_GATK4 = "/home/software/java/1.8.0_181/bin/java -jar -Xmx8g /home/software/gatk/4.1.3.0/gatk-package-4.1.3.0-local.jar";
$SOFT_GATK4 HaplotypeCaller -I ./K15_final.bam -O ./K15.g.vcf.gz -R "/home/ucsc/hg38_rm_alt_genome/bwa_idx/hg38_modify.fa" -ERC GVCF --min-base-quality-score 20 --sample-ploidy 2 --tmp-dir /home/tmp -L /home/ucsc/hg38_rm_alt_genome/realign_Target_hg38.bed --max-reads-per-alignment-start 0 --dont-use-soft-clipped-bases true
#使用GATK4 HaplotypeCaller ,输入比对结果K15_final.bam,物种参考基因组hg38_modify.fa,目标区域重排列realign_Target_hg38.bed,物种倍型信息,过滤参数等... 输出获得./K15.g.vcf.gz文件。
运行完成后每个样本都会获得对应的gVCF文件和索引:K15.g.vcf.gz、K15.g.vcf.gz.tbi
生成VCF文件
前面步骤是每个样本各自单独生成gVCF文件,这一步是将所有样本的gVCF文件汇总成一个包括所有样本突变信息的VCF文件。
本步骤要使用picard软件来实现。picard是基于java环境下的软件Picard Tools – By Broad Institute
合并后的vcf文件再进行过滤处理(其中有使用到bcftools软件)。
# GVCF 分型,由于分析较慢,故按照染色体拆分处理
#这里只展示chr1染色体上的分析,其他的染色体可以套上循环并行处理。
#需要提前对bed文件按染色体拆分获得$chr_1_bed(chr1的bed)
$SOFT_GATK4 CombineGVCFs -L $chr_1_bed -R "/home/ucsc/hg38_rm_alt_genome/bwa_idx/hg38_modify.fa" --variant Sample1.g.vcf.gz --variant Sample2.g.vcf.gz ...... -O ./chr_1_dir/chr1.sample.all.g.vcf.gz --tmp-dir /home/tmp
$SOFT_GATK4 GenotypeGVCFs -L $chr_1_bed -R "/home/ucsc/hg38_rm_alt_genome/bwa_idx/hg38_modify.fa" -V ./chr_1_dir/chr1.sample.all.g.vcf.gz -O ./chr_1_dir/chr1.sample.all.raw.vcf.gz --sample-ploidy 2 --tmp-dir /home/tmp
#这里的循环运行完就会获得1-22+X+Y染色体的chr*.sample.all.g.vcf.gz、chr*.sample.all.raw.vcf.gz文件
#把所有染色体的chr*.sample.all.raw.vcf.gz放入@split_vcfs 数组中。# VCF 合并
map{ $split_vcf_list .= "-I $_ " }@split_vcfs;
$SOFT_GATK4 MergeVcfs $split_vcf_list -O ./2.sample.all.raw.vcf.gz --TMP_DIR /home/tmp
#2.sample.all.raw.vcf.gz里存放的即 所有样本的所有染色体上的突变信息合并结果。# VCF 后续过滤、预处理
#这里的过滤思路一般都是针对SNP,INDEL,MIXED三种情况进行过滤(加标签)
# GATK4 SelectVariants
# GATK4 VariantFiltration
#过滤方法一般常见有两种:VQSR 过滤,HARD 硬过滤
#各种过滤后的结果汇总为sample.all.filter.vcf.gz#使用bcftools 对vcf的多态位点拆分,左校正(统一突变的记录格式)
/home/software/bcftools/1.9/bin/bcftools norm -m -both -f "/home/ucsc/hg38_rm_alt_genome/bwa_idx/hg38_modify.fa" -O z -o ./sample.all.final.vcf.gz ./sample.all.filter.vcf.gz
/home/software/bcftools/1.9/bin/bcftools index --tbi ./sample.all.final.vcf.gz运行完成后会获得所有样本汇总的VCF突变文件:sample.all.final.vcf.gz、sample.all.final.vcf.gz.tbi
这里的VCF文件包含了我们通过过滤筛选出的全部突变位点信息、每个样本在每个突变位点上的分型信息。是后续突变检测分析的基础。
如:围绕Plink软件,结合分组和表型经行snp关联分析、GWAS全基因组关联分析。(VCF文件格式可通过GATK、Plink、vcftools等软件转换成Plink文件格式)VCF和Plink转换
本篇后续是通过ANNOVAR等软降对位点进行注释,获取每个位点更为详细的注释信息,然后后续继续WES分析。
突变位点信息注释
前面获得的VCF文件主要包括两大块信息:
1:位点信息(染色体、位置、REF基因型、ALT基因型、INFO……)
2:每个样本在这个位点上的详细(REF分型信息、ALT分型信息、测序深度、分型质量……)
- 得到一系列变异数据,这些变异数据只是告诉我们在基因组的某个位置发生了一段序列的改变,至于这个改变会不会影响生物学功能,我们并不清楚。而注释就是将基因组的序列变异数据转化为我们更关心的生物学功能变化的信息。
- Annovar常被用在人类基因组的注释上,其实,它也可以对人类以外的基因组数据进行注释。比如,老鼠基因组的注释上。需要自己进行建立注释信息。
- ANNOVAR是一个perl编写的命令行工具,能在安装了perl解释器的多种操作系统上执行。允许多种输入文件格式,包括最常被使用的VCF格式。输出文件也有多种格式,包括注释过的VCF文件、用tab或者逗号分隔的text文件。
- ANNOVAR能快速注释遗传变异并预测其功能。类似的variants注释软件还有 VEP, snpEff, VAAST, AnnTools等等.
本流程将围绕Annovar软件来介绍突变位点注释
### ANNOVAR 的文件结构
|-- annotate_variation.pl # 主程序,功能包括下载数据库,三种不同的注释
|-- coding_change.pl # 可用来推断蛋白质序列
|-- convert2annovar.pl # 将多种格式转为.avinput的程序
|-- retrieve_seq_from_fasta.pl # 用于自行建立其他物种的转录本
|-- table_annovar.pl # 注释程序,可一次性完成三种类型的注释
|-- variants_reduction.pl # 可用来更灵活地定制过滤注释流程
|-- example # 存放示例文件
|-- humandb # 人类注释数据库ANNOVAR是一款对基因变异进行功能注释的软件,可以对多种生物的变异进行注释(包括 human genome hg18, hg19, hg38, 以及 mouse, worm, fly, yeast 等)。 给定一个包含染色体,起点,终点,参考核苷酸与观测核苷酸, ANNOVAR可以进行如下的功能注释:
- 基于基因的注释 Gene-based annotation:主要针对SNP或CNV是否引起蛋白编码改变进行注释,可以灵活选用 RefSeq genes, UCSC genes, ENSEMBL genes, GENCODE genes, AceView genes等多种不同来源的基因定义系统。
- 基于区域的注释 Region-based annotation:针对基因组某一特定区域的变异进行注释,例如44个物种的保守区域,预测的转录因子结合位点,GWAS hit, ENCODE H3K4Me1/H3K4Me3/H3K27Ac/CTCF sites,ChIP-Seq peaks, RNA-Seq peaks等
- 基于筛选的注释 Filter-based annotation:使用某一特定的数据库进行筛选注释,例如注释变异的rs id,1000基因组项目中的MAF,或是ExAC、gnomAD等,再例如SIFT/ PolyPhen/ LRT/ MutationTaster/ MutationAssessor/ FATHMM/ MetaSVM/ MetaLR 分数等。
annotate_variation.pl
annotate_variation.pl 模块有两种功能:一:是下载数据库;二:是三种注释方法(即上面提到的三种)
–downdb下载数据库
Arguments to download databases or perform annotations
--downdb download annotation database
--geneanno annotate variants by gene-based annotation (infer functional consequence on genes)
--regionanno annotate variants by region-based annotation (find overlapped regions in database)
--filter annotate variants by filter-based annotation (find identical variants in database)对于基于基因的注释的数据库已经在下好的 ANNOVAR package中了。如果要进行其他注释,需要按以下命令下载数据库到 ‘hg19/’ 目录里(这里路径是自定义的)
perl annotate_variation.pl --downdb --webfrom annovar --buildver hg38 refGene hg38/
perl annotate_variation.pl --downdb --buildver hg38 cytoBand hg38/
perl annotate_variation.pl --downdb --webfrom annovar --buildver hg38 1000g2014oct hg38/
perl annotate_variation.pl --downdb --webfrom annovar --buildver hg38 exac03 hg38/
perl annotate_variation.pl --downdb --webfrom annovar --buildver hg38 ljb26_all hg38/
perl annotate_variation.pl --downdb --webfrom annovar --buildver hg38 clinvar_20240611 hg38/
perl annotate_variation.pl --downdb --webfrom annovar --buildver hg38 avsnp156 hg38/
# --downdb 下载功能
# -webfrom annovar 从annovar库里下载;如果annovar库中没有,则不用写该选项,会从UCSC中下载
# -buildver 基因组对应版本
# refGene、cytoBand、1000g2014oct...... 数据库名称
# hg38/ 下载至该目录(数据库路径)
###-buildver的参数 会在hg38/路径下 生成对应文件,所以这两个要注意统一使用(下文提到的的脚本也是一样)- 这里下载的是几个通常用到的数据库:
- 0)refGene hg38基因组的refGene数据库(自带的是没有的,所以我下载了一个。)
- 1)cytoBand 是每个细胞间band(cytogenetic band)的染色体坐标信息 ,
- 2)1000g2014oct 是2014年10版,1000基因组项目(和ExAV 外显子集合联合一样,是公开、开放的数据库)里面供选择的等位基因频率信息
- 3)exac03 是0.3版外显子集合联合中报道过的variants.
- 4)ljb26_all 人类非同义SNPs的轻量级数据库及其在ResearchGate上的功能预测
- 5)clinvar_20240611 ClinVar是美国国家生物技术信息中心(NCBI)启动的公共、免费数据库。作为核心数据库,ClinVar数据库整合了十多个不同类型数据库、通过标准的命名法来描述疾病,同时支持科研人员将数据下载到本地中,开展更为个性化的研究。在遗传变异和临床表型方面,NCBI和不同的研究组已经建立了各种各样的数据库,数据信息相对比较分散,ClinVar数据库的目的在于整合这些分散的数据、将变异、临床表型、实证数据以及功能注解与分析等四个方面的信息,通过专家评审,逐步形成一个标准的、可信的、稳定的遗传变异-临床表型相关的数据库。
- 6)avsnp156 dbSNP数据库(version 156).
注意:
(i) 第二个命令中不包含 –webfrom annovar 选项, 因此是从the UCSC Genome Browser annotation database下载文件的;
(ii) –buildver hg38 选项是针对hg38这一版的基因组的;
(iii) 运行上面命令后,在 hg38/ 目录下会多几个以 hg38为前缀的文件。
–geneanno、–regionanno、–filter数据注释
perl annotate_variation.pl --geneanno -buildver hg38 sample.all.final.avinput hg38/使用annotate_variation.pl的基因注释功能(–geneanno )使用hg38/下面的(refGene)数据库,对我的输入数据sample.all.final.avinput(这个数据后面介绍) 进行注释。
注释获得了:sample.all.final.avinput.exonic_variant_function、sample.all.final.avinput.variant_function两个结果。
[wangzq@server0 annovar]$ ll -th
total 32M
-rw-rw-r-- 1 wangzq wangzq 978 Jul 11 2024 sample.all.final.avinput.log
-rw-rw-r-- 1 wangzq wangzq 4.1M Jul 11 2024 sample.all.final.avinput.exonic_variant_function
-rw-rw-r-- 1 wangzq wangzq 7.3M Jul 11 2024 sample.all.final.avinput.variant_function
drwxrwxr-x 3 wangzq wangzq 4.0K Jul 11 15:16 soft
-rw-rw-r-- 1 wangzq wangzq 4.9M Jul 11 10:00 sample.all.final.avinput
-rw-rw-r-- 1 wangzq wangzq 443K Jul 11 09:57 sample.all.final.vcf.gz.tbi
-rw-rw-r-- 1 wangzq wangzq 15M Jul 11 09:55 sample.all.final.vcf.gz–regionanno、–filter两种注释方法类似,这里不再介绍,在后面的table_annovar.pl 中会统一介绍。这里只列出help文档中的用法
Example: #download annotation databases from ANNOVAR or UCSC and save to humandb/ directory
annotate_variation.pl -downdb -webfrom annovar refGene humandb/
annotate_variation.pl -downdb -buildver mm9 refGene mousedb/
annotate_variation.pl -downdb -buildver hg19 -webfrom annovar esp6500siv2_all humandb/
#gene-based annotation of variants in the varlist file (by default --geneanno is ON)
annotate_variation.pl -geneanno -buildver hg19 ex1.avinput humandb/
#region-based annotate variants
annotate_variation.pl -regionanno -buildver hg19 -dbtype cytoBand ex1.avinput humandb/
annotate_variation.pl -regionanno -buildver hg19 -dbtype gff3 -gff3dbfile tfbs.gff3 ex1.avinput humandb/
#filter rare or unreported variants (in 1000G/dbSNP) or predicted deleterious variants
annotate_variation.pl -filter -dbtype 1000g2015aug_all -maf 0.01 ex1.avinput humandb/
annotate_variation.pl -filter -buildver hg19 -dbtype snp138 ex1.avinput humandb/
annotate_variation.pl -filter -dbtype dbnsfp35a -buildver hg38 ex1.avinput humandb/
annotate_variation.pl -filter -dbtype gnomad211_exome -buildver hg19 ex1.avinput humandb/
#-filter -buildver是参考基因组版本(名称) -dbtyp是注释用的数据库版本(名称)**/ 是注释数据库存放路。存放的数据humandb/hg38_snp138.txt
table_annovar.pl
table_annovar.pl脚本可以实现多个数据库一起注释。table_annovar.pl脚本的输入可以是 sample.all.final.avinput格式,也可以是sample.all.final.vcf.gz格式。
perl table_annovar.pl sample.all.final.avinput hg38/ -buildver hg38 -outfile myanno -remove
-protocol refGene,cytoBand,clinvar_20240611
-operation g,r,f- sample.all.final.avinput annovar标准格式的输入文件
- hg38/存放参考数据库的路径
- -buildver hg38 指定基因组版本,要和上面的存放数据库路径下数据库名字对应
- -remove 删除中间文件
- -protocol 选项后跟注释来源数据库的准确名称
- –operation选项后跟注释的类型:
g 表示基于基因的注释(gene-based annotation)
r 表示基于区域的注释(region-based annotation)
f 表示基于筛选子的注释( filter-based annotation) - -outfile 输出结果文件前缀
关键步骤:
- 1、确保注释数据库的名称正确并且是按你想要在输出文件中显示的顺序排列的;
- 2、确保 –operation指定的注释类型顺序和–protocol指定的数据库顺序是一致的;
- 3、确保每个protocal名称或注释类型之间只有一个逗号,并且没有空白。
perl table_annovar.pl sample.all.final.vcf.gz hg38/ -buildver hg38 -outfile myanno -remove
-protocol refGene,cytoBand,clinvar_20240611
-operation g,r,f
-vcfinput #指定输入是vcf文件(软件会自己生成**.avinput文件格式) 注释结果写在输出vcf文件的INFO列中。myanno.hg38_multianno.vcf 输出文件应该是一个VCF格式文件,
INFO那列以key=value 形式、分割成几个小区域.eg: Func.refGene=intronic;Gene.refGene=SAMD11. 每个键值对代表一个ANNOVAR注释信息。输出文件可以用为VCF格式文件设计的基因分析软件进一步处理。
myanno.hg38_multianno.txt 每一行代表一个variant 。用tab分隔,多余列为加上的注释信息,顺序按 –protocol选项所设定的注释类型argument。
convert2annovar.pl
convert2annovar.pl 脚本是将其他格式的文本转换为ANNOVAR专用的.avinput格式。
VCF文件转换为.avinput:
convert2annovar.pl -format vcf4old sample.all.final.vcf.gz --includeinfo --comment --outfile sample.all.final.avinput
#--includeinfo 保留VCF的INFO信息
#--comment 保留VCF的表头其他物种注释数据库
ANNOVAR数据库中只包含人类基因组已建好的转录本,不包含其他物种的。但软件是支持其他物种注释的,需要自行去建立数据库(retrieve_seq_from_fasta.pl)。本流程不涉及其他物种,这里就不做介绍。
至此,我们已经获得了全部样本的突变位点信息、每个样本在突变位点上的分型信息、每个突变位点的注释信息。
后续就是对获得的文本信息整理,一般推荐整理成database.snv、analysis.txt、annotation.txt三个文本格式
database.snv存放全部的突变位点(包括一些基础的注释信息):
chr1|12807|C 2 rs62635285 C T chr1 12807 DDX11L1 ncRNA_intronic CGCCCCTAGGGCTCTACGGGGCCGGCGTCTCCTGTCTCCTGGAGAGGCTT GATGCCCCTCCACACCCTCTTGATCTTCCCTGTGATGTCATCTGGAGCCC 9 chr1|12807|C|T
chr1|13079|C 2 rs78890234 C G chr1 13079 DDX11L1 ncRNA_intronic TGAAGGCTGTCAACCAGTCCATAGGCAAGCCTGGCTGCCTCCAGCTGGGT GACAGACAGGGGCTGGAGAAGGGGAGAAGAGGAAAGTGAGGTTGCCTGCC 9 chr1|13079|C|G
chr1|13418|G 2 rs75175547 G A chr1 13418 DDX11L1 ncRNA_exonic TGTGGAAGTTCACTCCTGCCTTTTCCTTTCCCTAGAGCCTCCACCACCCC AGATCACATTTCTCACTGCCTTTTGTCTGCCCAGTTTCACCAGAAGTAGG 9 chr1|13418|G|A
chr1|13649|G 2 rs879707275 G C chr1 13649 DDX11L1 ncRNA_exonic TGGAGTCCAGAGTGTTGCCAGGACCCAGGCACAGGCATTAGTGCCCGTTG AGAAAACAGGGGAATCCCGAAGAAATGGTGGGTCCTGGCCATCCGTGAGA 9 chr1|13649|G|C
chr1|14542|A 2 rs1045833 A G chr1 14542 WASH7P ncRNA_exonic CCCATGGAGCACAGGCAGACAGAAGTCCCCGCCCCAGCTGTGTGGCCTCA GCCAGCCTTCCGCTCCTTGAAGCTGGTCTCCACACAGTGCTGGTTCCGTC 9 chr1|14542|A|G
............
analysis.txt存放每个样本在位点上的分型信息(也包括一些质量打分):
chr1|69511|A G 1.0000 , , S_6_1:HOMA:G/G:0:159:159:99:5097:477:0,S_7_1:HOMA:G/G:0:78:78:99:2509:234:0,
chr1|930314|C T 1.0000 , S_6_1:HET:C/T:21:16:37:99:463:0:651,S_7_1:HET:C/T:20:17:37:99:508:0:587, ,
chr1|942402|C G 1.0000 , S_6_1:HET:C/G:6:6:12:99:141:0:147,S_7_1:HET:C/G:4:3:7:96:96:0:142, ,
chr1|942451|T C 1.0000 , , S_6_1:HOMA:C/C:0:10:10:30:321:30:0,S_7_1:HOMA:C/C:0:7:7:21:234:21:0,
chr1|948245|A G 1.0000 , , S_6_1:HOMA:G/G:0:56:56:99:2144:168:0,S_7_1:HOMA:G/G:0:44:44:99:1485:131:0,
chr1|953279|T C 1.0000 , , S_6_1:HOMA:C/C:0:178:178:99:6468:534:0,S_7_1:HOMA:C/C:0:153:153:99:5138:456:0,
chr1|953778|G C 1.0000 , , S_6_1:HOMA:C/C:0:30:30:90:1341:90:0,S_7_1:HOMA:C/C:0:42:42:99:1877:126:0,
chr1|953779|A C 1.0000 , , S_6_1:HOMA:C/C:0:30:30:90:1341:90:0,S_7_1:HOMA:C/C:0:42:42:99:1877:126:0,
chr1|962060|G A 1.0000 S_6_1:HOMR:G/G:169:0:169:99:0:120:1800, S_7_1:HET:G/A:81:79:160:99:2555:0:2364, ,
chr1|966748|C T 1.0000 , S_6_1:HET:C/T:32:32:64:99:975:0:956,S_7_1:HET:C/T:35:27:62:99:818:0:1120, ,
..........annotation.txt则存放除了基础注释信息以外的全部注释信息:
chr1|14599|T A 数据库名**** 注释信息****
chr1|14604|A G 数据库名**** 注释信息****
chr1|14930|A G 数据库名**** 注释信息****
chr1|69897|T C 数据库名**** 注释信息****
chr1|135982|A G 数据库名**** 注释信息****
chr1|732994|G A 数据库名**** 注释信息****
chr1|805887|T C 数据库名**** 注释信息****
chr1|817514|T C 数据库名**** 注释信息****
chr1|826577-826577|- T 数据库名**** 注释信息****
chr1|826893|G A 数据库名**** 注释信息****
..........遗传模式分析
结合样品的表型和突变信息可以很方便地定位疾病位点,因此,根据家系模式或疾病模式进行显/隐性遗传分析是疾病研究中的常见手段。此外,针对单基因遗传病还可以开展致病基因/位点的筛查研究, 比如家系连锁分析(家系样本数目大于20)、关联分析(样本数目大于50,包含对照样本)或者散发样本的共有突变定位等。
根据遗传遗传模式的过滤分析 ,不同的项目都有不同的过滤方法和过滤阈值,这里就不做深入讲解。(后续会根据一个实际例子单独出一篇文章介绍)