
fastQC
最常用的质控工具
二代测序下机的原始数据拿到手之后,第一步要做的事情就是看一看reads的质量。最常用的工具就是FastQC。
无论是RNA-seq、DNA-seq甚至三代测序的下机fastq数据都可以使用FastQC来进行质控。
fastQC可以从官网或者Conda下载,这里不再过多赘述
安装好之后可以“fastqc –help”查看帮助手册。fastQC功能非常单一,使用更简单。
其中有一个“–extract”参数,在pipeline中推荐使用。(正常情况输出结果是压缩包,使用此参数可自动解压。方便文件后续使用)
fastp
fastq文件质控过滤工具(还有一个过滤工具Trimmomatic,个人使用不如fastp,就不在做介绍)
fastp可以实现处理数据的一次性处理,包括过滤低质量,过滤adapter,截取reads, 全局剪裁,split分割大文件等操作。
支持长reads,也就是不仅仅适用二代测序,还支持三代测序长测序数据。
直接输出质控和统计报告,包括json格式和html格式。
fastp是陈实富老师在github上开源的软件,可以直接下载,可以使用”wget http://opengene.org/fastp/fastp”命令直接下载。
fastp软件的功能非常多,具体内容请根据软件自带的–help文档参考学习。
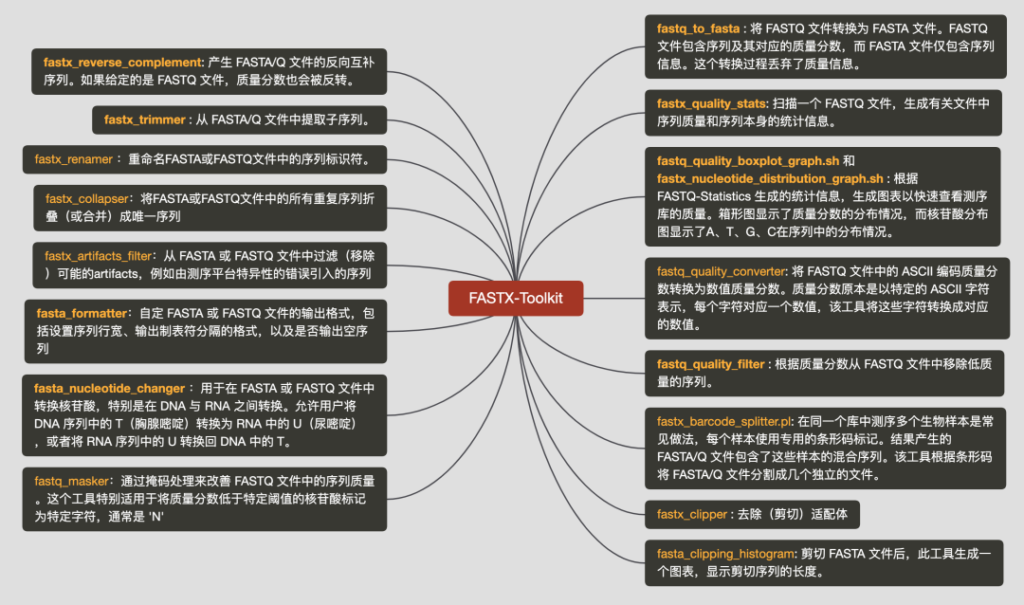
FASTX-Toolkit
FASTX-Toolkit是用于短读FASTA / FASTQ文件预处理的命令行工具的集合。包含多个原始reads处理的小命令工具。
可通过官网下载安装、Conda安装。
conda create -p 存放路径 fastx-toolkit
fastx_toolkit由一系列的命令组成,每个命令提供一个实用的小功能。在使用时需要注意以下几点:
- 不支持压缩格式的输入文件
- 不允许序列中存在N碱基,这样的序列会自动去除
- 可视化命令依赖gunplot软件和perl的GD模块
- 默认情况下认为fastq文件的碱基编码格式为phred64
Pingback:RNA-seq数据分析流程-Ⅰ – MyBioDatas