
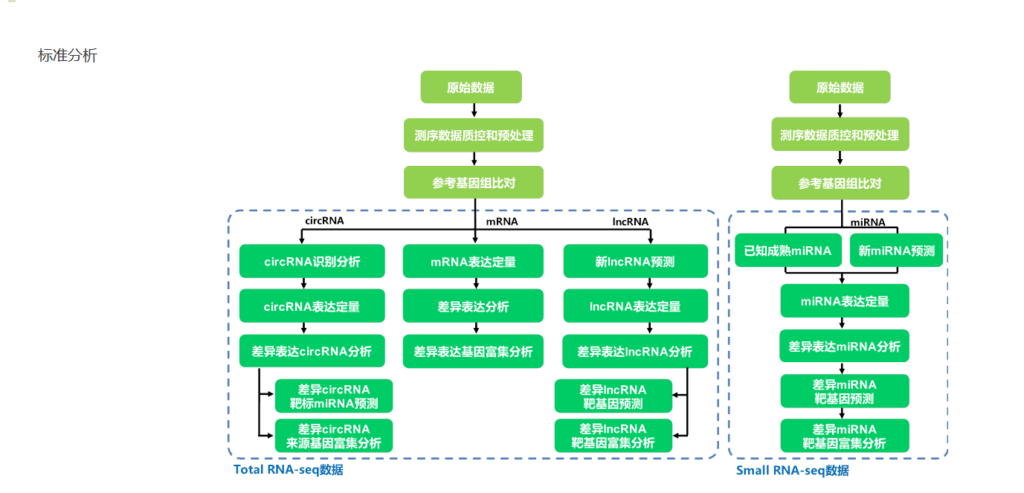
RNA-seq最常见的就是WTS转录组测序分析,其他的还有单细胞转录组、m6A(N6-腺苷酸甲基化)分析等。本篇讲以WTS(全转录组分析,包括了RNA测序分析+miRNA测序分析)来介绍整套分析流程。
下面是一整套WTS分析流程的思维导图,实验部分有两块,一个是RNA文库制备、一个是miRNA文库制备。制备文库一块属于湿实验部分,我完全不了解…
我们的分析是从fastq数据为起点开始的。(如果是网络下载的数据,可以参考SRA Toolkit获取)
流程图里包括了RNA分析(其中包括mRNA、lncRNA、circRNA)、miRNA分析、联合分析。
本文章只对mRNA、miRNA两个部分从脚本层面详细介绍。其他RNA部分只从思路层面介绍,帮助理解整个转录组分析框架。
RNA 质量控制
拿到数据的第一步就是质量控制,分为:原始数据质控、数据过滤、过滤后数据质控。
原始数据质控
数据质控使用fastQC软件、seqtk软件 对原始数据进行碱基数量、质量统计分析。
[wangzq@server6 wangzq]$ /home/software/fastqc/0.11.5/fastqc -t 10 --extract -q -o ./K15 --dir ~/tmp/ /home/project/K15_R1.fastq.gz /home/project/K15_R2.fastq.gz
Too many tiles (>500) so giving up trying to do per-tile qualities since we're probably parsing the file wrongly
Too many tiles (>500) so giving up trying to do per-tile qualities since we're probably parsing the file wrongly
# -t --threads 指定线程
# --extract 输出文件将在同一目录下进行解压缩,非交互式运行情况下建议添加此参数
# -q --quiet 禁止stdout上的所有进度消息,只报告错误。
# -o --outdir 指定输出文件路径
# -d --dir 用于生成报告图像时写入的临时文件。如果未指定,默认为系统临时目录。
#################################################################################################
[wangzq@server6 wangzq]$ /home/software/seqtk/seqtk fqchk /home/project/K15_R1.fastq.gz > ./R1.quality.txt
[wangzq@server6 wangzq]$ /home/software/seqtk/seqtk fqchk /home/project/K15_R2.fastq.gz > ./R2.quality.txt
#seqtk fqchk 碱基质量统计 ,使用统计结果配合R软件绘制碱基分布图,错误率图等基础信息图片。数据过滤
数据过滤使用fastp软件 对raw数据进行去接头、低质量碱基过滤。
[wangzq@server6 wangzq]$ /home/software/fastp/fastp -w 10 -l 40 -3 \
--in1 /home/project/K15_R1.fastq.gz --in2 /home/project/K15_R2.fastq.gz \
--out1 ./K15_final_R1.fastq.gz --out2 ./K15_final_R2.fastq.gz
--html ./fastp.html --json ./fastp.json
# -w --thread 指定线程
# -l --length_required 指定最小长度
# -3 --cut_tail 质控时,滑动窗口从3'端开始
# 如需要还可以添加 --adapter_sequence --adapter_sequence_r2 参数指定接头序列。
# --in1 --in2 输入的PE reads文件
# --out1 --out2 过滤后输出的PE reads文件(自行指定名字,我这里是在名字上加了final标签)
# --html --json 指定质控的输出文件位置 这两个输出文件包含了详细的过滤情况的记录。
# ......fastp软件有很多参数,这里不一一列举。过滤后数据质控
过滤后数据质控和原始数据一样使用fastQC软件、seqtk软件 对原始数据进行碱基数量、质量统计分析。
[wangzq@server6 wangzq]$ /home/software/fastqc/0.11.5/fastqc -t 10 --extract -q -o ./K15_final --dir ~/tmp/ ./K15_final_R1.fastq.gz ./K15_final_R2.fastq.gz
Too many tiles (>500) so giving up trying to do per-tile qualities since we're probably parsing the file wrongly
Too many tiles (>500) so giving up trying to do per-tile qualities since we're probably parsing the file wrongly
# -t --threads 指定线程
# --extract 输出文件将在同一目录下进行解压缩,非交互式运行情况下建议添加此参数
# -q --quiet 禁止stdout上的所有进度消息,只报告错误。
# -o --outdir 指定输出文件路径
# -d --dir 用于生成报告图像时写入的临时文件。如果未指定,默认为系统临时目录。
#################################################################################################
[wangzq@server6 wangzq]$ /home/software/seqtk/seqtk fqchk ./K15_final_R1.fastq.gz > ./final_R1.quality.txt
[wangzq@server6 wangzq]$ /home/software/seqtk/seqtk fqchk ./K15_final_R2.fastq.gz > ./final_R2.quality.txt
#seqtk fqchk 碱基质量统计 ,使用统计结果配合R软件绘制碱基分布图,错误率图等基础信息图片。RNA 基因组比对(Alignment)
比对是指将我们的fastq数据比对到该物种的参考基因组上去,获得比对结果的bam/sam 文件。这里我们选择RNA比对软件STAR作为核心工具,配合stringtie、cuffmerge、samtools 共同使用。STAR比对之前需要对参考基因组进行建库索引(物种参考基因组和GTF建库,这是一个非常耗时的工作,一般都会提前准备好。具体建库步骤在以后会具体介绍STAR软件)
cuffmerge是转录组工具cufflinks套装中的一个小工具,用于merge GTF文件。
[wangzq@server6 wangzq]$ /home/software/star/2.7.10b_conda/bin/STAR --readFilesIn ./K15_final_R1.fastq.gz ./K15_final_R2.fastq.gz \
--genomeDir /home/database/star/hg19/ref_genome.fa.star.idx \ #这里就是提前准备好的参考物种数据库索引
--readFilesCommand gunzip -c \
--outFileNamePrefix ./K15_ --outTmpDir ./K15_tmp --outTmpKeep All \
--chimSegmentMin 12 --chimJunctionOverhangMin 8 --chimOutJunctionFormat 1 #这几个参数是控制输出K15_Chimeric.out.junction,这个文件用于后续的融合基因分析(fusion)
#STAR软件的比对有非常非常多的参数可以调控,控制线程、控制输出文件format、控制存储占用、控制输出文件属性(attr)、控制Overlap相关参数......
#这里不对此做详细介绍,根据自己需要,参考STAR的document自行设置。
#比对的输出结果是bam文件,重点结果:./K15_Aligned.sortedByCoord.out.bam
[wangzq@server6 wangzq]$ /home/software/samtools/1.7/samtools/index ./K15_Aligned.sortedByCoord.out.bam
#对star比对的bam结果建立samtools索引
#这里生成的bam即用于后续定量使用。
###############
#####下面的gtf操作都是对 新转录本预测 做准备工作,如果只考虑基础分析,可以忽略这部分。(这里主要使用的是stringtie 的不加 -e 的模式来预测)
[wangzq@server6 wangzq]$ /home/software/stringtie/1.4.1/stringtie ./K15_Aligned.sortedByCoord.out.bam -o ./K15.gtf \
-G /home/database/star/hg19/mRNA_lncRNA.gtf
#这里G参数给参考GTF文件,如果是RNA测序分析,一般都是STAR建库时使用的GTF文件(但一般情况下从各个网站下载的GTF文件都会有不同的文件格式风格,还需要自行对格式处理)
#获得的./K15.gtf 文件已经包含了样本的初始定量结果(FPKM和TPM和cov)
#一般转录组都会分析多个样本,这里要进行合并gtf的工作
[wangzq@server6 wangzq]$ for sample in K15.gtf K16.gtf K17.gtf ;do echo ./${sample} >> ./list.txt ;done #准备工作把多个样本的gtf写到同一个txt中
[wangzq@server6 wangzq]$ /home/software/cufflinks/2.2.1/cuffmerge -g /home/database/star/hg19/mRNA_lncRNA.gtf -o ./transcripts ./list.txt
#这样,多个样本比对的gtf结果就被merge到一个汇总的GTF文件中了。用于后续新转录本预测。RNA 比对质量控制
比对质控控制即 比对效果的汇总,对整个比对过程与结果给出一个可视化的统计分析。本步骤主要使用picard软件和RSeQC软件来实现。picard是基于java环境下的软件。(RSeQC是RNA-seq高通量测序质控及可视化工具包,可直接通过pip或者Conda安装)
还有Python、R软件统计可视化。
/home/software/java/1.8/bin/java -jar /home/software/picard/2.18.29/picard.jar CollectRnaSeqMetrics \
I=./K15_Aligned.sortedByCoord.out.bam \
REF_FLAT= /home/software/picard/hg19/hg19.refflat.txt \
RIBOSOMAL_INTERVALS= /home/software/picard/hg19/hg19.ribosomal_intervals.txt \
O=./K15_metrics.tsv STRAND_SPECIFICITY=SECOND_READ_TRANSCRIPTION_STRAND
# REF_FLAT、RIBOSOMAL_INTERVALS 是picard CollectRnaSeqMetrics 分析必须的文件,可提前用gtf文件、picard软件、ucsc的gtfToGenePred软件生成
# REF_FLAT 文件包换GTF里除了rRNA之外的信息,文件一行表示一个转本。文本从第一列开始每列表示:基因名、转录本名、染色体、链方向、转录起始位点、转录终止位点、CDS起始位点、CDS终止位点、外显子数量、外显子起始位点、外显子终止位点
# RIBOSOMAL_INTERVALS 用于存储基核糖体RNA(rRNA)的区间信息。
#REF_FLAT、RIBOSOMAL_INTERVALS两个文件具体信息可参考picard CollectRnaSeqMetrics 的document文档。
#./K15_metrics.tsv 即比对质量统计结果,可根据此结果绘制分布饼图、柱状图等,这里不再详细介绍RPKM_saturation.py
read_distribution.py
read_duplication.py
geneBody_coverage.py
#上面几个是RSeQC包里比对质量控制常用的脚本,输入文件是比对bam文件(K15_Aligned.sortedByCoord.out.bam)、bed12格式的文件(必须使用bed12格式,自行提前准备,一行一个转录本,内容包含转录本信息及转录本里的外显子信息)mRNA lncRNA 表达量定量
表达量定量是使用前面比对获得的bam文件,基于stringtie软件、stringtie软件配套的prepDE.py工具,ballgown函数包,得到样本的定量矩阵。
分析思路是逐个样本获取定量结果,然后在汇总成一个完整的矩阵。(如果是mRNA、lncRNA同时比对的,还需要对比对结果进行拆分)
简单理解为:基于bam文件 通过stringtie软件获得了gtf数据、ballgown数据 两套结果;
用prepDE.py工具搭配gtf数据获得count_matrix矩阵;用ballgown函数包搭配ballgown数据获得FPKM矩阵。
[wangzq@server6 wangzq]$ /home/software/stringtie/1.4.1/stringtie -p 10 -G /home/database/star/hg19/mRNA_lncRNA.gtf \
-o ./K15/profile_K15.gtf -b ./K15 -e ./K15_Aligned.sortedByCoord.out.bam
# 对每个样本进行定量,如果有多个样本,每个样本都需要单独的定量,获得对应的profile_K15.gtf文件
# 这里使用stringtie软件搭配参考GTF文件、-e参数限制输出。
# 使用-e参数后,StringTie的输出将仅限于那些在参考注释文件中列出的转录本,任何新的转录本都不会被预测和输出。
# -b 参数用于指定输出 Ballgown 输入表文件(*.ctab)的目录路,ballgown数据用于后续生成FPKM矩阵。
[wangzq@server6 wangzq]$ for sample in K15 K16 K17 ;do echo -e '${sample}\t./${sample}/profile_${sample}.gtf' >> ./list.txt ;done
# 准备工作把单独的定量的profile_K15.gtf 文件写入到./list.txt文本中,用于后续样本合并定量。
[wangzq@server6 wangzq]$ /home/software/python/2.7.15/bin/python /home/software/stringtie/1.4.1/prepDE.py \
-i ./list.txt -g ./merge_gene_count_matrix.csv -t ./merge_transcript_count_matrix.csv
#ballgown数据使用R语言的ballgown函数包可整理出fpkm结果(merge_transcript_fpkm.txt、merge_gene_fpkm.txt),这里不再列出代码了。
# merge_gene_count_matrix.csv
# merge_gene_fpkm.txt
# merge_transcript_count_matrix.csv
# merge_transcript_fpkm.txt
# 我们后续的差异分析、富集分析都是基于count这一套结果进行的。因为这个例子我使用的是RNA分析,包含了mRNA、lncRNA合并定量(使用的参考GTF都是mRNA_lncRNA.gtf )。所以后续的还需要把mRNA 、lncRNA拆分开来,在进行后续分析。
如果前面使用的是mRNA或者lncRNA单独的GTF来比对、定量的 ,这个步骤可以跳过。
这里的拆分具体内容是:根据GTF文件中涉及到的“gene_id”、“transcript_id”来提取子矩阵。
(大致思路分为两步,1:获取GTF中全部的gene_id、transcript_id;2:用这两个id,从汇总的矩阵中过滤获得目标结果)
拆分完成之后mRNA、lncRNA分别会获得gene_count_matrix、gene_fpkm_matrix、transcript_count_matrix、transcript_fpkm_matrix 四个表达量矩阵。
拆分完成之后,可以进行一些简单的描述性统计分析、结果可视化。
例如fpkm的density密度曲线绘制、基于R语言DESeq2包的标准化矫正数据、基于校正后数据绘制PCA主成分图、基于校正后数据绘制correlation各样本之间的相关性图……
最后可以根据需要对定量文件加上注释信息。
基因定量可以加上基因类型、染色体起始终止、转录因子……
转录本定量可以加上转录本类型、所属基因、染色体起始终止、转录本上的外显子信息、CDS信息……
mRNA 表达量差异分析
转录组的差异分析常见的有三种:DESeq2、edgeR、limma。
- DESeq2:是一种基于负二项分布广义线性模型的差异表达分析方法,适用于RNA-seq数据。DESeq2能够处理测序深度影响,并通过标准化因子区分基因表达差异。
- edgeR:是基于负二项分布的模型,它通过估计离散度来估计生物学变异系数(BCV),进而拟合线性回归模型的参数。edgeR适用于计数数据,能够进行精确的差异表达基因检测。
- limma:limma包原本用于微阵列数据分析,但通过voom转换后,也可以用于RNA-seq数据的差异分析。limma-voom方法在处理计数数据时表现出较好的性能,尤其是在样本量较小的情况下。
本流程选择使用DESeq2对gene_count_matrix、transcript_count_matrix进行差异分析。除了表达量矩阵外还必须知道差异分析的样本分组信息。
DESeq2是一个R语言的package包,可用过Bioconductor或者Conda安装。(主要使用DESeqDataSetFromMatrix、DESeq、counts、results等几个个函数)
R语言的具体脚本这里不列出了。
在DESeq2差异分析的结果里都会有baseMean、log2FoldChange、pvalue、padj、stat等统计量。
一般使用“log2FoldChange的绝对值>1”并且“pvalue<0.05”为阈值进行过滤,过滤出的结果即差异基因(转录本)
差异分析做完之后,根据分析结果进行结果可视化,可绘制correlation图片、MA图片、valcano图片、heatmap图片、PCA图片
图片绘制基本都是基于R语言的ggplot2系列函数包生成。
mRNA 差异基因的功能(GO KEGG)富集
功能富集分析,作为生物信息学数据分析的重要组成部分,旨在揭示生物分子功能、生物学过程以及信号通路的潜在机制。基因本体论(GO)、京都基因与基因组百科全书(KEGG)和基因集富集分析(GSEA)是三种常用的功能富集分析方法,它们在揭示基因功能、探索疾病机制以及药物研发等方面发挥着重要作用。
本步骤将使用前面获得的差异基因作为分析的起点,分别介绍GO、KEGG、GSEA 这三类富集分析。
这一步主要使用R语言的“clusterProfiler ”包 搭配AnnotationHub的orgdb数据库 来完成富集分析。(不同的物种都有对应的orgdb数据库)
GO富集分析
GO富集分析可以帮助我们了解基因在生物学过程(BP)、细胞组分(CC)和分子功能(MF)上的富集情况。
使用clusterProfiler包中的enrichGO函数进行GO富集分析:
GO富集分析一般使用的都是ENTREZID格式的基因编号,所以需要把SYMBOL格式的基因名转换成ENTREZID,(根据该物种的orgdb数据库,使用bitr函数可轻松转换)enrichGO函数的结果是类变量的保存格式,可直接对类结果使用plotGOgraph函数绘制BP、CC、MF的富集分析。三个结果再分别取topN的差异显著结果汇总,绘制总的barplot图。
KEGG富集分析
KEGG富集分析可以揭示基因在信号通路和代谢途径中的富集情况。
使用clusterProfiler包中的enrichKEGG函数进行KEGG富集分析:
KEGG富集分析一般使用的都是ENTREZID格式的基因编号,所以需要把SYMBOL格式的基因名转换成ENTREZID,(根据该物种的orgdb数据库,使用bitr函数可轻松转换),enrichKEGG函数会自动连接到KEGG的在线数据库进行注释,因此需要确保网络连接正常。enrichKEGG还需要organism参数,是分析物种的缩写名称,可以通过:’http://www.genome.jp/kegg/catalog/org_list.html’ 查看。
GSEA富集分析
GSEA(基因集富集分析)是一种无需设定阈值来区分上调或下调基因的方法,它利用所有的基因进行分析,从而提供更全面和深入的生物信息学见解。
GSEA的输入文件是全部基因(包括差异显著和不显著)和相应的logFC。
我对于GSEA的理解:使用全部的基因来进行类似上面的GO、KEGG分析。使用的是gseGO函数、gseKEGG函数
mRNA 蛋白互作网络(PPI)
蛋白质-蛋白质相互作用(protein-protein interaction, PPI)是指两个或两个以上的蛋白质分子通过非共价键形成蛋白质复合体(protein complex)的过程。
系统分析大量蛋白在生物系统中的相互作用关系,对了解生物系统中蛋白质的工作原理,了解疾病等特殊生理状态下生物信号和能量物质代谢的反应机制,以及了解蛋白之间的功能联系都有重要意义。在生物医药领域有助于从系统的角度研究疾病分子机制、发现新药靶点等等。
这里的PPI分析基于STRING数据库、R语言的STRINGdb包 进行分析,获得蛋白(基因)之间的相互关系。并使用igraph和ggraph可视化蛋白互作网络图。
基于STRING的PPI分析需要 差异基因列表、分析的物种ID编号(从STRING官网可查)、对应物种的string本地数据库(ppi_stringdb)
mRNA 可变剪切 新转录本 融合基因
可变剪切
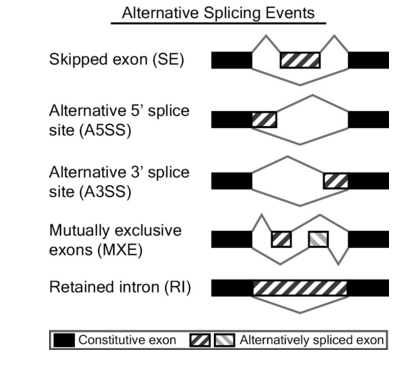
基因在转录过程中,不同的剪接方式产生不同的mRNA。RNA测序可以较好的捕获mRNA的可变剪切信息。采用rMATS软件进行可变剪切分析
可变剪切的分类:
- 外显子缺失 (Exon skipping);
- 可变的5’端剪切 (Alternative 5’ splicing);
- 可变的3’端剪切 (Alternative 3’ splicing);
- 互斥外显子 (Mutually exclusive exons);
- 内含子包含 (Intron retention)。
#准备包含样本比对参考基因组的bam文件路径的文本文件
echo K15.bam,K16.bam > b1.txt
echo C17.bam,C18.bam > b2.txt
#运行RMATS软件
rmats.py --b1 b1.txt --b2 b2.txt --gtf /home/database/star/hg19/mRNA_lncRNA.gtf --od ./alternative_splicing \
--nthread 10 --tstat 10 --readLength 100 -t paired
# --b1 --b2 分别指定实验组和对照组的bam文件路径
# --gtf 指定参考基因组的GTF文件
# --od 指定输出目录
# paired 表示数据是双端测序
# --nthread 指定使用的线程数
# --readLength 指定reads的长度
RMATS软件提供了 rmats2sashimiplot 工具,可以将rmats的输出结果可视化,绘制sashimi plot。具体详细操作参考RMATS软件的document。
新转录本
在前面基因组比对的时候我们就有提到,stringtie 的不加 -e 的模式来进行预测,后续我们得到的./transcripts/merged.gtf文件中既包含了参考GTF中已知的转录组,还包含了一定量的预测转录本。从预测的转录本逆推到参考基因组上的序列信息,再使用DIAMOND把这些序列比对到NR/NT数据库,比对上的结果即预测出的新转录本。
(这里的介绍非常粗糙,因为我也没有实际操作运行过。)
融合基因
基因融合(Gene fusion)是指两个基因的全部或一部分的序列相互融合为一个新的基因的过程,所得到的这个基因被称之为融合基因(Fusion gene)。 融合基因所表达的融合蛋白在功能上通常有所变异。采用STAR-Fusion鉴定融合基因。基于软件STAR-Fusion预测得到的全部潜在的融合基因信息。(这里的介绍非常粗糙,因为我也没有实际操作运行过。)
至此,转录组分析的mRNA分析部分全部介绍完了。下面粗略介绍一下其他RNA相关分析。
lncRNA分析介绍
前面在表达量定量时我们对lncRNA也获取了 gene_count_matrix、gene_fpkm_matrix、transcript_count_matrix、transcript_fpkm_matrix 四个表达量矩阵。
类似mRNA的表达量差异分析(使用gene、transcript的countMatix),获得差异的lncRNA基因-ID、lncRNA转录本-ID。
用差异lncRNA基因-ID、lncRNA转录本-ID 做目标基因映射(由lncRNA的定义可知,lncRNA本身不编码蛋白质,是作用在mRNA、miRNA上从而起到调控功能的)。
(这里需要提前准备LncTar数据库,用于lncrna_mrna的映射关系)。获得映射后的差异基因列表,即可进行功能富集分析(GO KEGG),方法也是和mRNA分析类似。
后续也可以进行lncRNA的可变剪切、新转录本分析。
miRNA 分析介绍
miRNA 是采用miRDeep2软件,将各样本的small RNA序列与miRBase数据库中对应物种的miRNA前体及成熟体进行比对,同时结合近缘物种的同源miRNA序列, 比如RNAfold等RNA二级结构,比对得到近缘的已知miRNA及其二级结构。
miRNA下机fastq与miRBase数据库中对应物种的miRNA前体及成熟体进行比对,比对上的结果即known miRNA;没比对上的再采用miRDeep2软件 预测novel miRNA。
这样分别获得known_mirna_expressed、novel_mirna_expressed 两个表达量矩阵。有此结果即可进行差异分析。再采用目标基因映射 的办法,获得miRNA对应的基因,这些即具有差异的基因,就可以进行功能富集分析(GO KEGG),方法也是和mRNA分析类似。
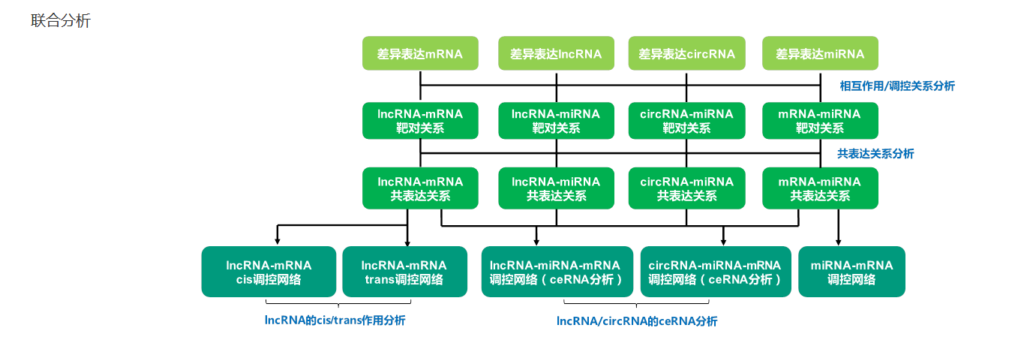
联合分析(Coexpression)
联合分析即上面的多种RNA的差异分析结果做联合分析,主要采用R语言的ggplot2、igraph和ggraph进行可视化蛋白互作网络图。
这里不再做过多介绍,详情可参考R语言的可视化绘图教程。